补充基础知识
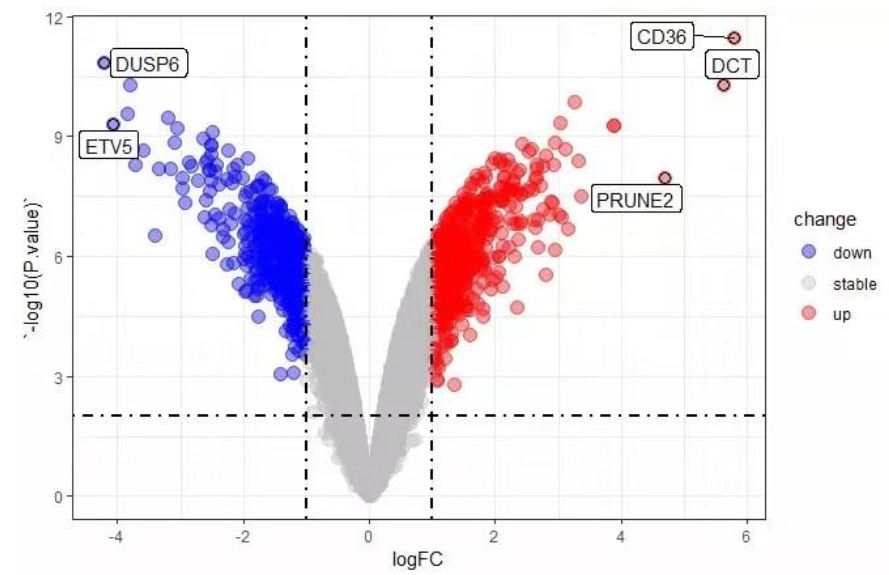
logFC
**FC(Foldchange):**处理组平均值/对照组平均值
**logFC:**Foldchange取log2
logFC基本都是个位数,一般作为横坐标出现在火山图中
- logFC>0, treat>control,基因表达量上升
- logFC<0, treat<control,基因表达量下降
- 通常所说的上调、下调基因是指表达量显著上升/下降的基因
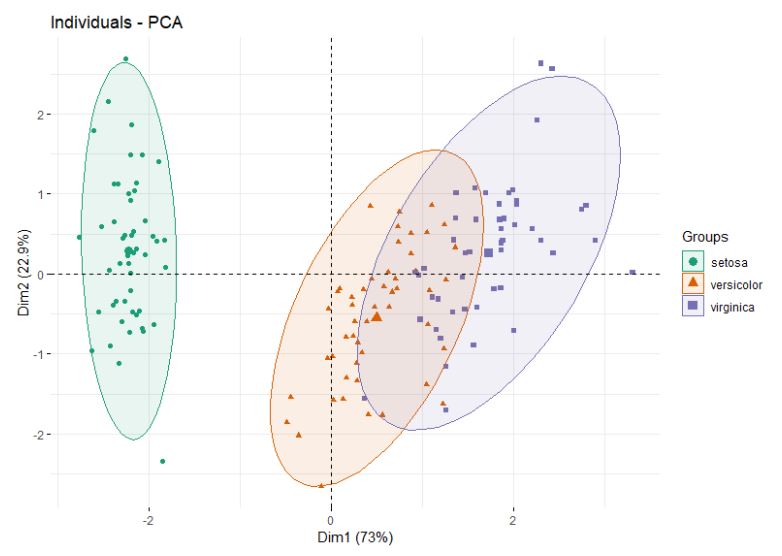
主成分分析(PCA)
主成分分析,旨在利用降维的思想, 把多指标转化为少数几个综合指标(即主成分)。
根据这些主成分对样本进行聚类,代表样本的点在坐标轴上距离越远,说明样本差异越大。
PCA只提供现象,无法解释现象发生的原因
GEO数据实战
- 数据来源在GEO网页上可以看到,array和high throughput sequencing
- 本次练习使用的数据均来自于芯片数据(array),不讨论转录组数据,注意甄别
研究背景
本次练习用到的数据来自于GEO数据,GSE号码为GSE56649.
Ye H, Zhang J, Wang J, Gao Y et al. CD4 T-cell transcriptome analysis reveals aberrant regulation of STAT3 and Wnt signaling pathways in rheumatoid arthritis: evidence from a case-control study. Arthritis Res Ther 2015 Mar 22;17:76. PMID: 25880754
类风湿关节炎(RA)是一种全身性自身免疫性疾病,其潜在的分子机制尚不清楚。此前,CD4 t细胞芯片研究只关注关节炎患者。我们的目的是比较活跃的RA和健康对照组CD4 T细胞的分子谱。
本次研究从13名活动的RA患者和9名健康个体中分离高度纯化的外周血CD4 T细胞,进行RNA提取和Affymetrix微阵列杂交。
数据获取
gse_number = "GSE56649" #设置num eSet <- getGEO(gse_number, destdir = '.', getGPL = F) #下载数据至本地工作空间 # 取列表中对象 eSet = eSet[[1]] class(eSet) - [1] "ExpressionSet" - attr(,"package") - [1] "Biobase" # 提取矩阵数据 exp <- exprs(eSet)
截取部分数据如下展示,列名为样本,行名为基因探针名字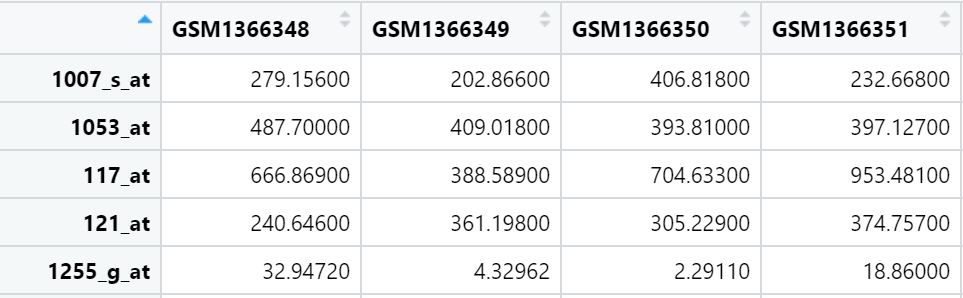
#查看数据是否有缺失、错误、负值 dim(exp) - [1] 54675 22 #判断是否需要log处理值 boxplot(exp) #生成箱线图
这是我们提取数据绘制的箱线图,可见箱子被压在底部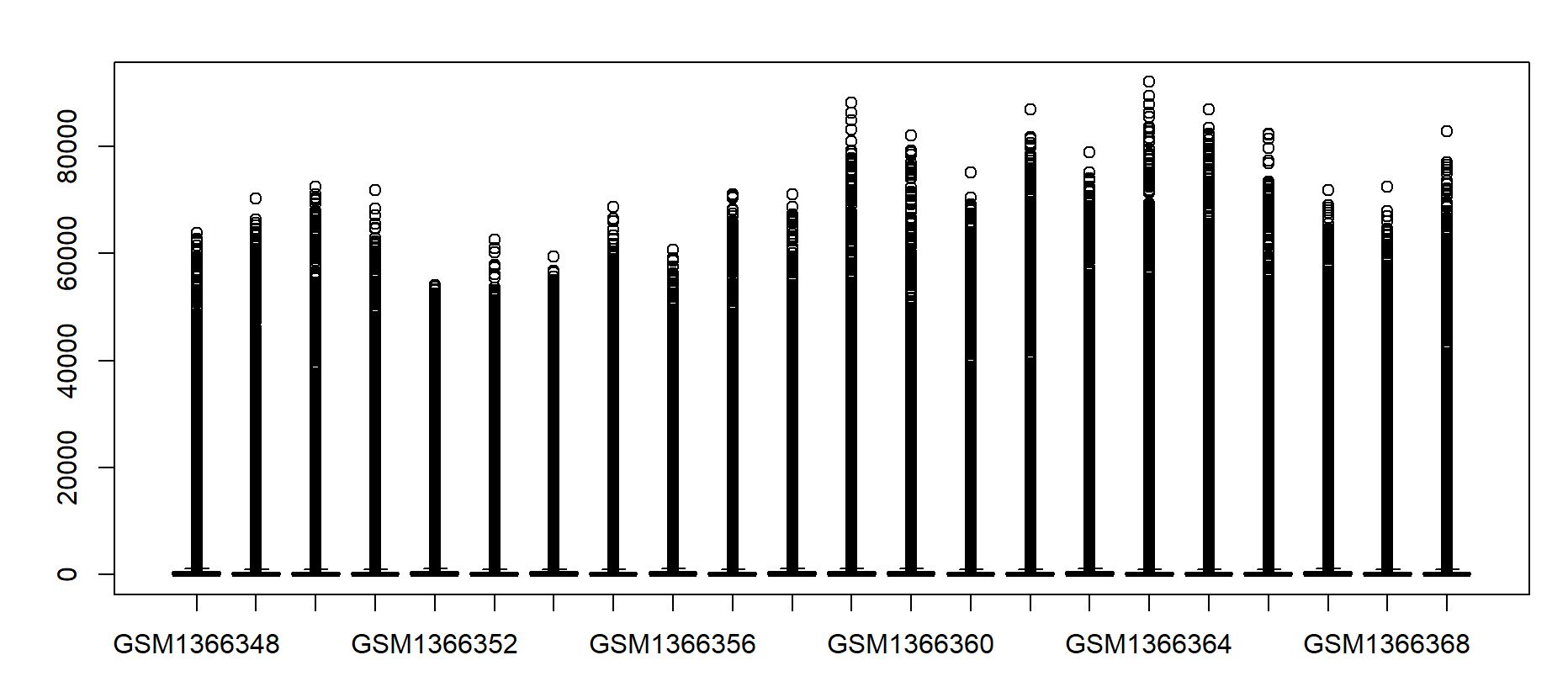
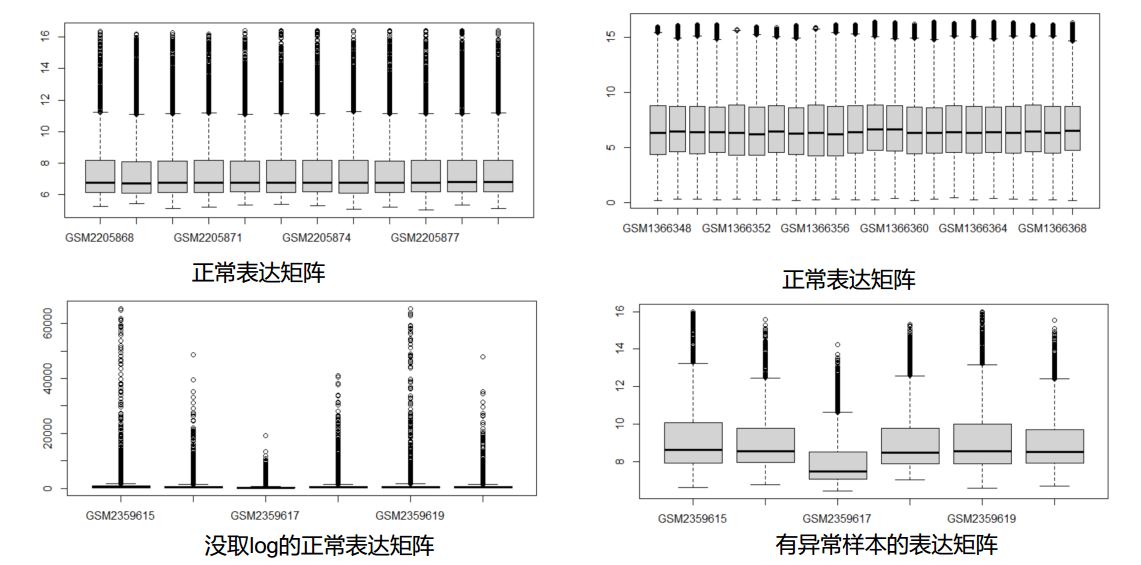
#将数据转换,约定俗成+1 exp = log2(exp+1) limma::normalizeBetweenArrays(exp) #如果存在很大的异常值,可以用该函数校正
#提取临床信息 pd <- pData(eSet) #让exp列名与pd的行名顺序完全一致 p = identical(rownames(pd),colnames(exp));p if(!p) exp = exp[,match(rownames(pd),colnames(exp))] #提取芯片平台编号 gpl_number <- eSet@annotation;gpl_number save(gse_number,pd,exp,gpl_number,file = "step1output.Rdata") 保存数据提取第一步
分组和探针注释
分组
rm(list = ls())
load(file = "step1output.Rdata")
library(stringr)
#分组.1 截取关键词
Group = pd$`disease state:ch1`
#分组.2 自己设置
Group = c(rep("RA",times=13),
rep("control",times=9))
Group = rep(c("RA","control"),times = c(13,9))
#分组.3 使用字符串的函数获取分组
Group=ifelse(str_detect(pd$source_name_ch1,"control"),
"control",
"RA")
# 将获取的分组变量,转化为factor变量,并且指定参考变量
Group = factor(Group,levels = c("control","RA"))
探针注释的获取
#捷径
library(tinyarray)
find_anno(gpl_number)
ids <- AnnoProbe::idmap('GPL570')
#方法1 BioconductorR包(最常用)
gpl_number
#http://www.bio-info-trainee.com/1399.html
if(!require(hgu133plus2.db))BiocManager::install("hgu133plus2.db")
library(hgu133plus2.db)
ls("package:hgu133plus2.db")
ids <- toTable(hgu133plus2SYMBOL)
head(ids)
# 方法2 读取GPL网页的表格文件,按列取子集
##https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL570
if(F){
library(GEOquery)
#注:表格读取参数、文件列名不统一,活学活用,有的表格里没有symbol列,也有的GPL平台没有提供注释表格
b = read.table("GPL570-55999.txt",header = T,
quote = "\"",sep = "\t",check.names = F)
colnames(b)
ids2 = b[,c("ID","Gene Symbol")]
colnames(ids2) = c("probe_id","symbol")
ids2 = ids2[ids2$symbol!="" & !str_detect(ids2$symbol,"///"),]
}
# 方法3 官网下载注释文件并读取
##http://www.affymetrix.com/support/technical/byproduct.affx?product=hg-u133-plus
# 方法4 自主注释
#https://mp.weixin.qq.com/s/mrtjpN8yDKUdCSvSUuUwcA
save(exp,Group,ids,gse_number,file = "step2output.Rdata")
如果出现一个探针对应多个基因-非特异性探针,可以去除